C. Michael DiPersio, PhD
Areas of Study
Normal and pathological tissue remodeling
Education
- Brown University1991PhD
- University of Massachusetts, Dartmouth1985BS
Research
As the major cell adhesion receptors for the extracellular matrix (ECM), integrins mediate ‘inside-out’ and ‘outside-in’ signaling that regulates the cell’s ability to both sense and promote changes in the tissue microenvironment. The DiPersio lab uses a variety of genetic, cell biological, and transcriptomic approaches to investigate how the laminin-binding integrin, α3β1, regulates cell functions that govern normal and pathological tissue remodeling. We apply these approaches to both cutaneous wound healing models and cancer models (skin and breast). This dual approach allows us to explore how functions of α3β1 that are tightly controlled in wound healing are dysregulated in carcinogenesis,building on the long held notion put forth by Harold Dvorak in 1986 that “tumors are like wounds that do not heal”. Our long-term goal is to develop therapeutic strategies that target α3β1 to inhibit cancer progression and metastasis, or to treat pathological wound healing (e.g., chronic wounds, hypertrophic scars).
Coordinated roles of epidermal integrins in wound healing and cancer.
To exploit integrins as therapeutic targets, we need to improve our understanding of how different integrins function coordinately. In collaboration with Dr. Livingston Van De Water’s group, we developed integrin knockout mice and mouse keratinocyte cell lines to investigate the coordinated functions of two epidermal integrins, α3β1 and α9β1. We determined that epidermal α3β1 is essential for several aspects of wound healing, including regeneration of the basement membrane and paracrine signaling to stromal cells that promotes wound angiogenesis (endothelial cells) or contraction (fibroblasts/ myofibroblasts). Moreover, we determined that α9β1 suppresses certain α3β1 functions, thereby acting as a temporally regulated “brake” on α3β1 during later stages of wound re-epithelialization when basement membrane assembly is complete, and during vascular regression when angiogenesis is no longer required.
Unlike healed wounds, tumors do not display vascular regression.We have determined that α9β1 expression is repressed in tumor cells during skin tumorigenesis, which likely promotes continued tumor growth by relieving the brake on α3β1 and allowing its pro-tumorigenic/pro-angiogenic functions to persist (see below). Indeed, forced expression of α9β1 in tumorigenic keratinocytes reduced their α3β1-dependent growth in vivoin a subcutaneous tumor model. Moreover, bioinformatic analysis of transcriptome data from human head and neck squamous cell carcinoma revealed that epigenetic silencing of theITGA9gene (which encodes the α9integrin subunit) may allow tumor cells to evade the tumor-suppressive effects of α9β1.
Roles of integrin α3β1 on tumor cells in regulation of the tumor microenvironment (TME).
A major focus of our research is on how α3β1 expressed on tumor cells regulates cancer progression through inter-cellular communication and modulation of the TME. For these studies we generated a genetic mouse model wherein α3β1 can be ablated in tumor keratinocytes through topical application of tamoxifen following 2-step chemical (DMBA/ TPA) skin tumorigenesis. Deletion of α3β1 from skin tumors leads to their rapid regression over 2 weeks, which is accompanied by dramatic changes in the TME that include increased apoptosis of stromal cells, reduced macrophages, and reduced fibulin-2 in the ECM. Comparative mass spec analysis of conditioned medium from immortalized keratinocytes that express or lack α3β1 revealed substantial differences in secreted proteins, including many that mediate ECM remodeling or stimulate stromal cells. Gene set enrichment analysis showed that a core subset of this α3β1-dependent secretome is enriched in human squamous cell carcinomas with high expression of ITGA3(the gene that encodes the α3integrin subunit). Moreover, RNA in situ hybridization of tumors from our in vivomodel showed that gene expression for two such proteins, fibulin-2 and macrophage colony-stimulating factor 1 (CSF1), was α3β1-dependent. Ongoing studies are focused on identifying the mechanisms through which α3β1 regulates the keratinocyte secretome that promotes a tumor-supportive TME.
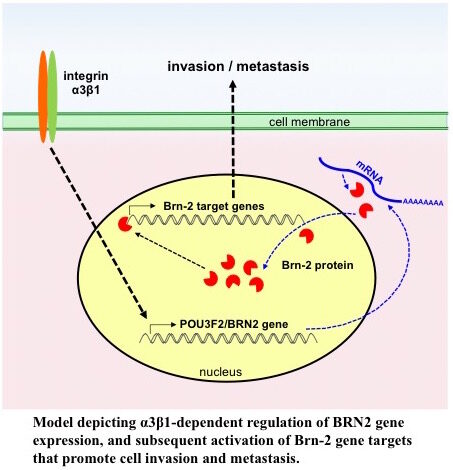
Roles for integrin α3β1 in regulating gene expression programs that promote cancer.
Another focus of our work is on molecular pathways through which α3β1 regulates gene expression that drives cancer progression. Through application of transcriptomic approaches (e.g., Affymetrix gene arrays, RNAseq) to our cell culture models, we discovered that deletion or suppression of α3β1 leads to global changes in gene expression and/or mRNA processing (e.g., splicing, 3’-UTR length). To identify mechanisms of this regulation we are focusing on specific α3β1-dependent gene targets in our different cancer models (see below).
Transcriptional gene regulation:
We used an Affymetrix gene array to identify changes that occur in the transcriptome of the triple-negative MDA-MB-231 breast cancer cell line when α3β1 is suppressed using shRNA. The transcription factor Brain-2 (Brn-2/Oct-7/N-Oct3/POU3F2) was among the top 15 transcripts that were α3β1-dependent. Subsequent functional studies showed that α3β1-mediated induction of Brn-2promotes invasion in vitro and metastatic lung colonization in vivo, and that exogenous Brn-2 partially restored invasion to cells in whichα3β1 was suppressed. Analysis of RNAseq data from patients with basal-like (i.e., triple-negative) breast cancer revealed that high BRN2expression correlates with high ITGA3expression and poor survival, supporting a pro-cancer role for the α3β1-Brn-2 axis. Ongoing studies are focused on identifying α3β1-regulated genes that are potential Brn-2 targets, including the genes that encode Cox-2 (PTGS2) and the extracellular protease Reelin (RELN).
Post-transcriptional gene regulation:
We have used our different cancer models to show that α3β1 promotes the expression of two pro-tumorigenic/pro-angiogenic genes, Cox-2 (PTGS2) and matrix metalloproteinase-9 (MMP9), by controlling alternative mRNA processing that determines susceptibility to mRNA degradation pathways. In the first model, RNAi-mediated suppression of α3β1 in breast cancer cells leads to alternative splicing of Cox-2 mRNA, thereby causing retention of an intron that harbors premature termination codons and targets the mRNA transcript for nonsense-mediated decay (NMD). In the second model, genetic deletion of α3β1 in immortalized keratinocytes leads to use of an alternative polyadenylation (APA) site in the MMP-9 gene to generate an extended 3’-UTR harboring several AU-rich elements (AREs) that destabilize the mRNA transcript. A common theme in both models is that α3β1 maintains expression of Cox-2 or MMP-9 mRNA by promoting the exclusion of regulatory elements from the transcript that would otherwise target it for a mRNA degradation pathway. Transcriptome profiling indicated that α3β1-dependent mRNA splicing or APA extends to many other genes. Future studies will include using our in vivo tumor models to investigate how α3β1-dependent mRNA processing contributes to cancer progression and metastasis.
Publications
View C. Michael DiPersio's articles on the National Institute of Health's PubMed website.
