Areas of Study
Disproportionate Inflammatory Responses
Education
- University of Buenos Aires2007PhD
- University of Buenos Aires1999BS/MS
Research
Our mission is to find novel therapeutics to treat tissue damage and organ dysfunction caused by disproportionate inflammatory responses. We aim to achieve that by identifying the most critical mechanisms of the pathophysiology that lead to chronic and excessive inflammation.
When inflammation goes awry
A cytokine-driven acute increase in vascular permeability is an essential feature of the inflammatory response leading to tissue repair and pathogen clearance. However, sustained vascular leakage due to prolonged or exacerbated inflammation leads to organ damage, lasting sequelae and increased mortality. Multiple edemagenic factors are known to participate in the initial changes in endothelial permeability via well-defined mechanisms. The mechanism mediating the acute (minutes to hours) responses to proinflammatory cytokine signaling are well-described: phosphorylation of tyrosines in adherens junction proteins and an increase in actin-myosin contractility are two main mechanisms leading to a quick loss of endothelial intercellular adhesion and thus an increase in vascular permeability. Despite its importance in severe and chronic inflammation, little is known about the changes in the endothelium leading to a sustained (lasting hours to days) vascular barrier breakdown. Understanding these long-term mechanisms may prove crucial to allow us to directly target vascular leakage and minimize tissue damage, thus reducing the rates of mortality and chronic sequelae of excessive edema.
Endothelial signaling leading to edema – a role for STAT3
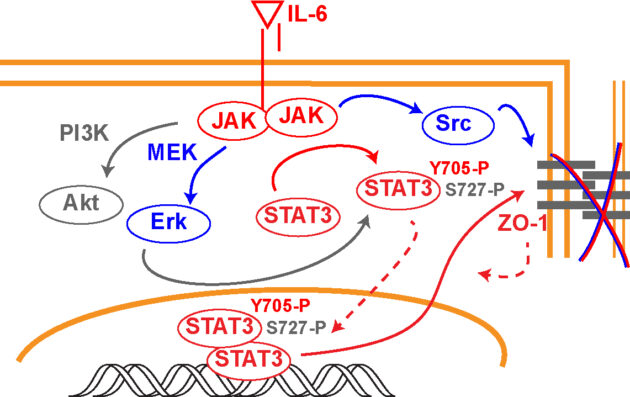
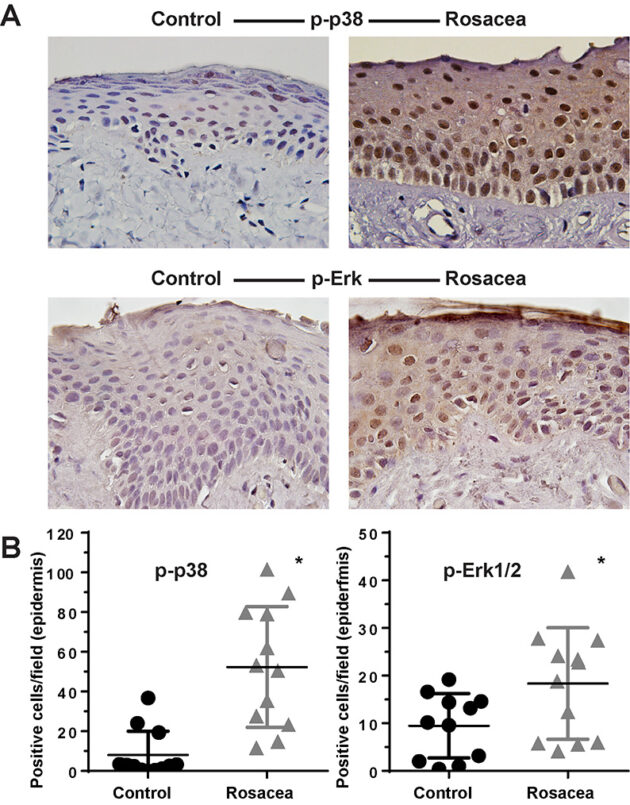
Innate immunity in rosacea
Edema in uveitis
Uveitis is a leading cause of blindness, accounting for approximately 10% of all cases of loss of sight. Uveitis is classified as anterior uveitis if it affects the iris or ciliary body, intermediate uveitis if presenting inflammation of the vitreous, or posterior uveitis when affecting the retina or choroid. Panuveitis refers to manifestations affecting most of the uvea. While rare in otherwise healthy individuals, uveitis can occur secondary to multiple causes, ranging from an infectious agent to autoimmune diseases. However, up to 50% of the cases remain idiopathic4. According to a study assessing insurance claims from over 4 million Americans, non-infectious uveitis accounts for over 90% of the cases. The retinal inflammation can cause macular edema, and in some cases choroid neovascularization, greatly impairing vision. A steroid treatment may cause immediate anti-inflammatory relief, but sustained use in chronic manifestations leads to the development of cataracts and glaucoma, thus limiting the therapeutic usefulness of these drugs. Anti-tumor necrosis factor (TNF-α) biologics, commonly used as treatment for multiple forms of arthritis for over a decade, showed efficacy to treat non-infectious uveitis and received FDA approval for its use in adults with non-infectious intermediate, posterior and panuveitis in 2016. Other FDA-approved biologics show also promising therapeutic potential for uveitis. Interleukin-6 (IL-6) is dramatically increased in the sera, aqueous and vitreous of uveitis patients. Further, some case studies and small clinical trials suggest that the use of IL-6 antagonists can alleviate the symptoms and improve vision in patients. These findings could be reproduced in animal models, where intravitreal injection of an anti-IL-6 attenuated experimental autoimmune uveitis. Studies performed in mice suggest that these biologics act at least in part by blocking Th17-mediated retinal inflammation. Consistently, loss of STAT3 in CD4+ T Cells prevented the development of experimental autoimmune uveitis in mice. Besides this insight, little is known about the mechanism of disease and the involvement of IL-6-induced pathways in disease progression. Moreover, any potential role for IL-6 signaling in the development of uveitic macular edema is currently unknown. Our current research is using transgenic mice to assess how the IL-6/STAT3 signaling axis regulates the development of macular edema during uveitis.
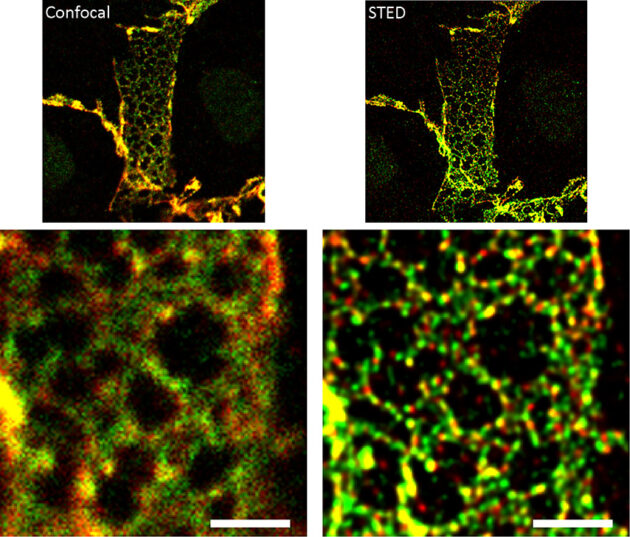
The endothelial adherens junction structure
Endothelial cells regulate the passage of fluids, macromolecules, and cells from the blood stream to the surrounding tissues. Endothelial permeability and transendothelial migration (TEM) are critical during acute and chronic inflammation, shock, atherosclerosis, acute lung distress syndrome, and primary tumor growth and metastasis. The EC is not only centrally involved in this process, but also readily exposed to circulating drugs and humanized antibodies, thus making it a perfect target for novel drug development. Hence, it is essential to understand its mechanisms of regulation in order to design better therapeutic strategies. Endothelial intercellular adhesion, and thus permeability, is tightly regulated by a number of vasoactive factors and is critical to determine the onset and extent of an inflammatory response. The endothelial AJ is comprised of the classical cadherin VE-cadherin and the catenins p120, β-catenin and plakoglobin. ECs contain AJ complexes along the cell-cell border in close association with a cortical actin ring, supporting strong cell-cell adhesion and resistance to the shear stress induced by the blood flow. Endothelial AJ can form very distinct structures within the cells, and their relative composition and function is currently unknown. Essentially, while we know the molecular partners of stable adherens junctions, little is known about the dynamic formation of these structures and why endothelial cells may show different AJ structures. The classical structure, termed here “linear” junction, is observed along the lines of cell-cell interactions in close association with the cortical actin ring. Linear junctions may occur together with “reticular” junctions, which are observed at regions of cell-cell overlap. These junctions are named after the distinct network of cadherins and catenins observed throughout the overlapping region. Linear AJ colocalize with tight junction markers, while reticular AJ do not. Further, linear junctions are less mobile than reticular junctions, suggesting that linear AJ may be more mature, stronger adhesion sites. Determining quantitatively the molecular composition of each structure is critical to be able to understand how endothelial cells interact and what is the role for each adherens junction structure. The lab is using several super-resolution strategies, including Airyscan and STED, and live tracking (mEOS2) in combination with AJ component mutants to better understand how the AJ forms and disassembles. The role for actin remodeling and focal adhesion components is also being studied.
Publications
View Alejandro P. Adam's articles on the National Institute of Health's PubMed website.